Fundamentals of organic chemistry
Introduction to organic chemistry
- Organic chemistry is the field of chemistry that studies carbon-based compounds. Carbon atoms have four valence electrons so they can form four bonds to other atoms. Carbon can undergo catenation, the process by which many identical atoms are joined together by covalent bonds, producing straight-chain, branched, or cyclic structures. Organic chemistry is therefore a wide and varied field of study.
Homologous series
- A homologous series is a series of compounds that can be grouped together based on similarities in their structure and reactions.
- A homologous series has the same general formula which varies from one member to another by one CH2 (methylene) group.
- The alkenes and alkynes are two more hydrocarbon homologous series that contain carbon– carbon double and triple bonds, respectively.
- Homologous series that contain functional groups can also be described by a general formula and also show similar physical and chemical properties within the series.
Successive members of a homologous series differ by a –– CH2 –– group
- Consider the following compounds of carbon and hydrogen where the carbons are all bonded by single covalent bonds.
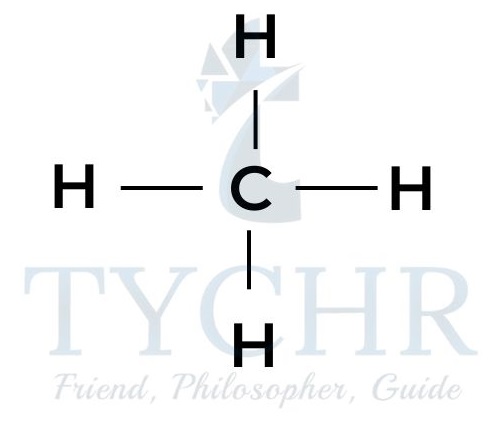
methane 
ethane 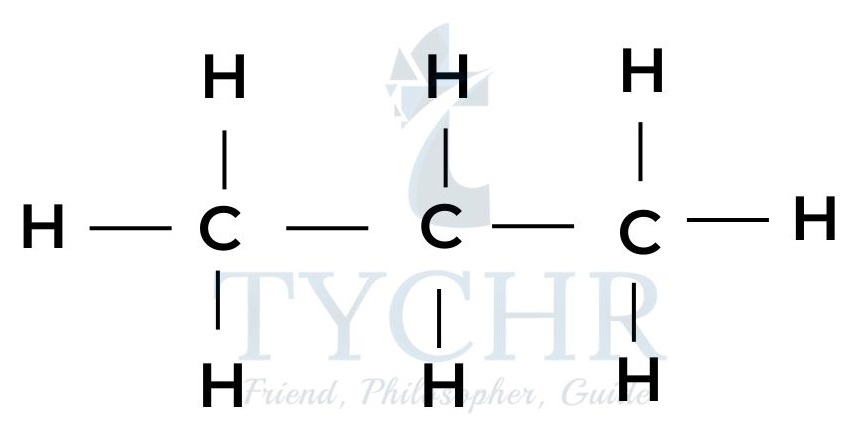
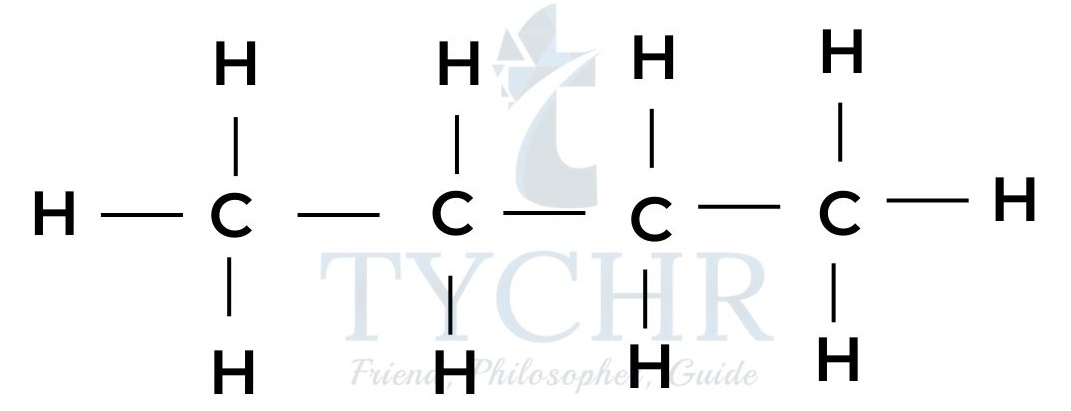
propane 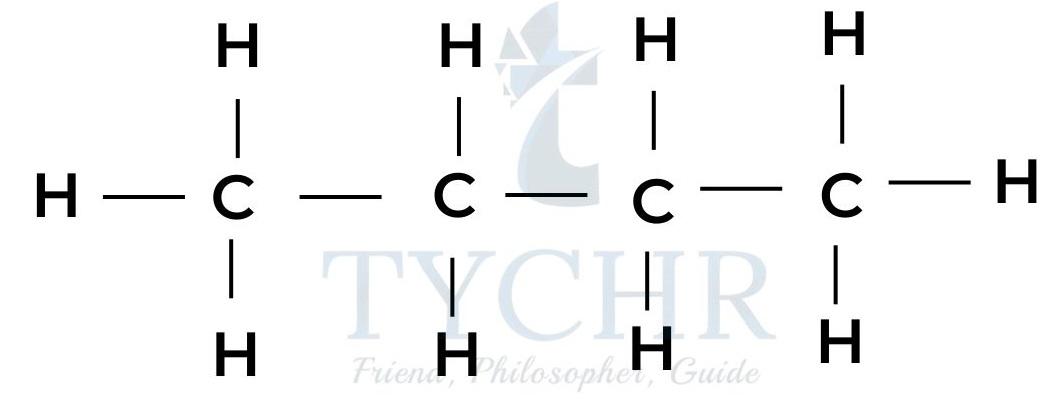
butane - These are members of a homologous series known as the alkanes. It can be seen that neighbouring members differ from each other by –– CH2 ––.
- This same increment applies to successive members of each homologous series, and means that the molecular mass increases by a fixed amount as we go up a series.




Members of a homologous series can be represented by the same general formula
- The four alkanes shown above can all be represented by the general formula CnH2n+2.
- Other homologous series are characterized by the presence of a particular functional group, and this will be shown in the general formula for the series.
- For example, the homologous series known as the alcohols possess the functional group –– OH as shown below.




Summary
- The main features of a homologous series are as follows:
- Successive members of a homologous series differ by a ——CH2—— group.
- The same general formula can be used to represent members of a homologous series..
- The members of a homologous series show a gradation in their physical properties.
- The members of a series have similar chemical properties.
Formulas for organic compounds: empirical, molecular, and structural
- The structure of an organic compound may be represented in several different ways providing varying levels of information.
- Empirical formulas represent the simplest ratio of atoms present in a molecule.
- The actual number of atoms in a molecule is described in the molecular formula.
- Both these types of formula offer little or no information about the possible structure of larger, more complex molecules.
- Structural formulas take three forms: full, condensed, and skeletal.
- Full structural formulae are two-dimensional representations showing all the atoms and bonds, and their positions relative to one another in a compound.
- In a condensed structural formula, all the atoms and their relative positions are represented but the bonds are omitted.
- A skeletal formula is the most basic representation of the structural formula where the carbon and hydrogen atoms are not shown but the end of each line and each vertex represents a carbon atom
Nomenclature for organic compounds
- The international authority on chemical nomenclature is the International Union of Pure and Applied Chemistry (IUPAC).
- The name of a chemical substance needs to provide enough information to signpost the class of compound from which the chemical is derived, including any substituents and functional groups present. The name has a number of parts that describe the compound (figure 1).

- The alkanes form the backbone of the IUPAC rules for naming organic compounds.
Structural isomers: different arrangements of the same atoms
- The molecular formula of a compound shows the atoms that are present in the molecule, but gives no information on how they are arranged. Consider for example the formula C4H10.
- There are two possible arrangements for these atoms that correspond to different molecules with different properties.
butane boiling point -0.5 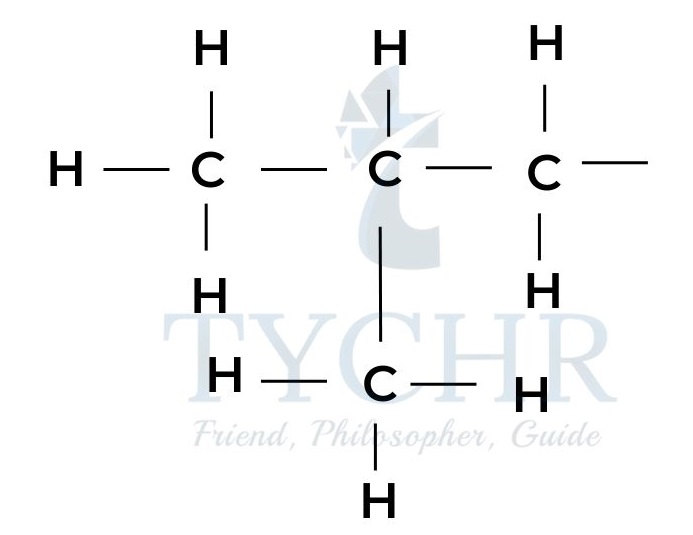
2-methylpropane boiling point -11.7 - Such molecules, having the same molecular formula but different arrangements of the atoms, are known as structural isomers.


Primary, secondary, and tertiary compounds
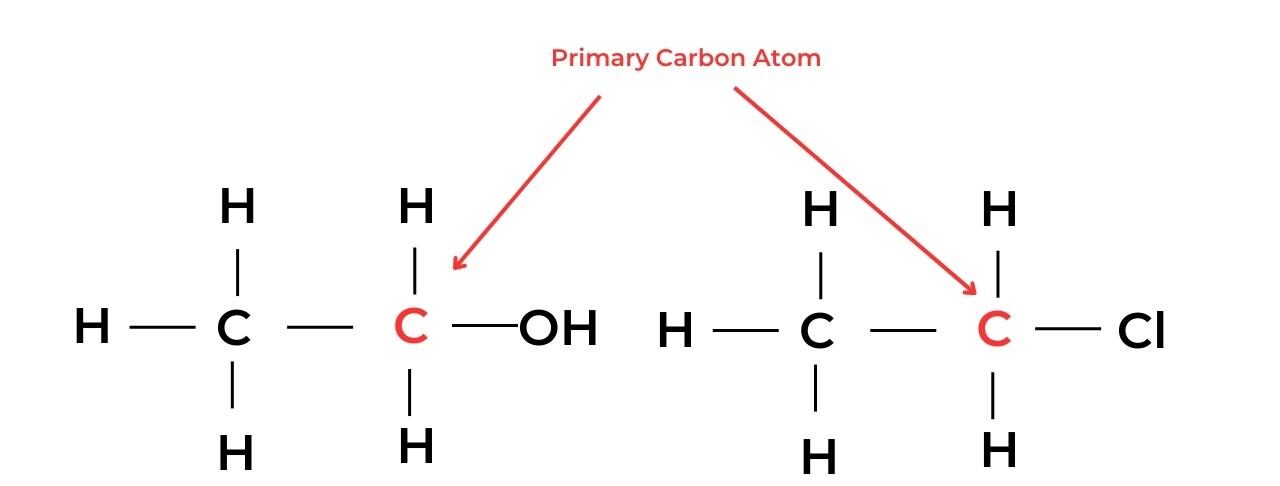
- A primary carbon atom is attached to the functional group and also to at least two hydrogen atoms. Molecules with this arrangement are known as primary molecules.
- For example, ethanol, C2H5OH, is a primary alcohol and chloroethane, C2H5Cl, is a primary halogenoalkane.
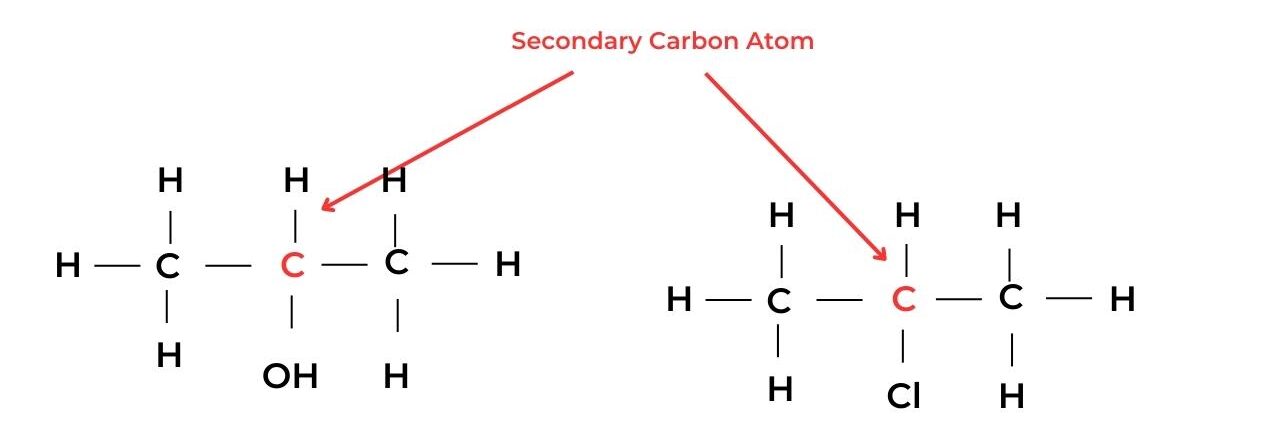
- A secondary carbon atom is attached to the functional group and also to one hydrogen atom and two alkyl groups. These molecules are known as secondary molecules.
- For example, propan-2-ol, CH3CH(OH)CH3, is a secondary alcohol, and 2-chloroethane, CH3CHClCH3, is a secondary halogenoalkane.
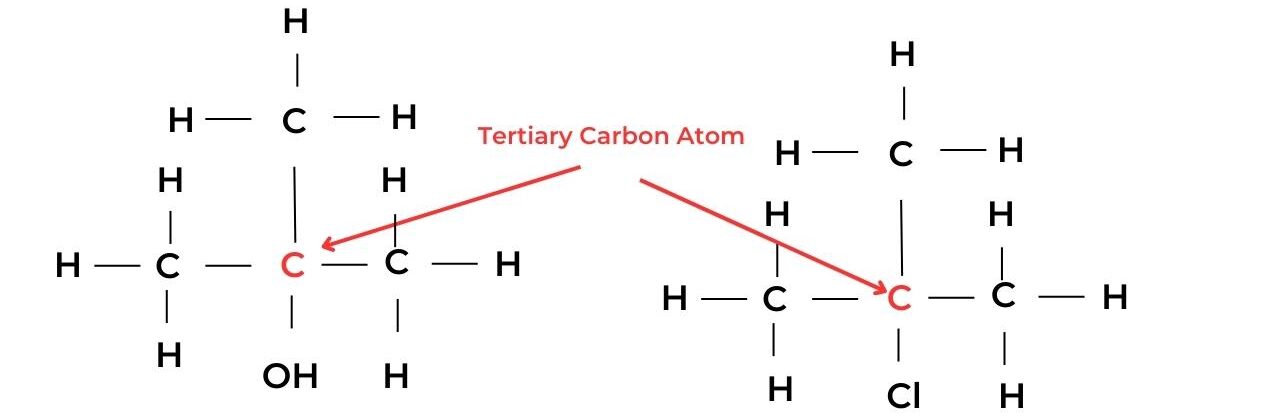
- A tertiary carbon atom is attached to the functional group and is also bonded to three alkyl groups and so has no hydrogen atoms. These molecules are known as tertiary molecules.
- For example, 2-methylpropan-2-ol, C(CH3)3OH, is a tertiary alcohol, and 2-chloro-2-methylpropane, C(CH3)3Cl, is a tertiary halogenoalkane.
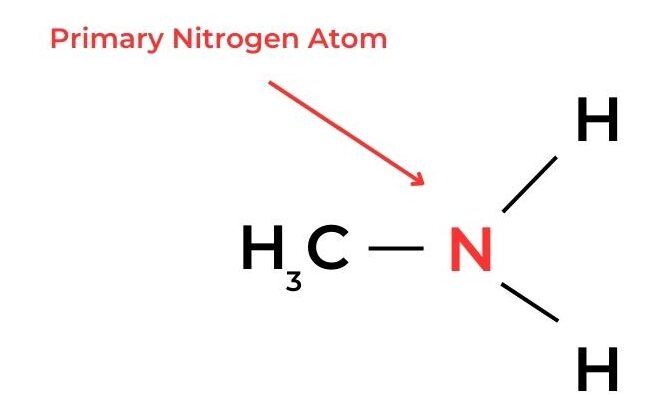
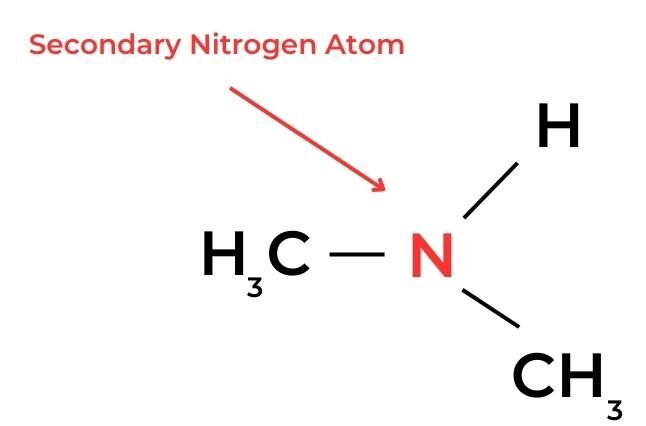
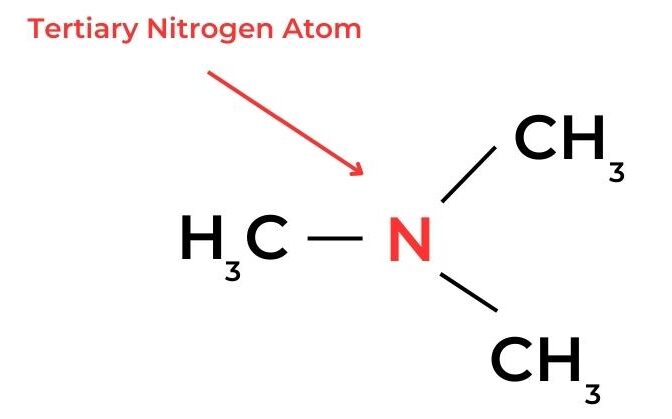
- In amines, a similar classification can be applied according to the number of alkyl groups and hydrogen atoms bonded to the nitrogen atom.






Arenes
- Arenes are a class of compounds that are derived from benzene, C6H6. They form a special branch of organic compounds known as the aromatics, which have properties somewhat distinct from other organic compounds, which are known as aliphatics. Arenes contain the phenyl functional group:

C6H5
Functional group chemistry
Alkanes
- Alkanes are the simplest hydrocarbons.
- With low bond polarity and strong covalent carbon–carbon bonds (bond energy 346 kJ mol-1) and carbon–hydrogen bonds (bond energy 414 kJ mol-1), they are relatively inert.
- However, alkanes do undergo some important reactions.
The combustion of alkanes
- Alkanes are commonly used as fuels, releasing large amounts of energy in combustion reactions.
- Alkanes undergo complete combustion in the presence of excess oxygen. This highly exothermic reaction produces carbon dioxide and water.
- Carbon dioxide has a significant environmental impact, contributing to global warming. Petrol or gasoline is a mixture of hydrocarbons with octane present in the highest proportion.
C8H18(l) + 12 1 /2 O2(g) → 8CO2(g) + 9H2O(g) ∆H = -5470 kJ
The halogenation of alkanes
- Alkanes are relatively inert, and chemists often work to activate alkanes to increase their reactivity. One way of achieving this is to halogenate the alkane.
- Common reactions studied in organic chemistry include substitution, addition, and elimination. Substitution is the replacement of individual atoms with other single atoms or with a small group of atoms. In an addition reaction two molecules are added together to produce a single molecule, while elimination is the removal of two substituents from the molecule.
- Alkanes can undergo free-radical substitution and elimination to form unsaturated alkenes and alkynes. Alkenes and alkynes can undergo all three types of reaction listed above.
Free-radical substitution
- The term free-radical refers to a species that is formed when a molecule undergoes homolytic fission: the two electrons of a covalent bond are split evenly between two atoms resulting in two free-radicals that each have a single electron:
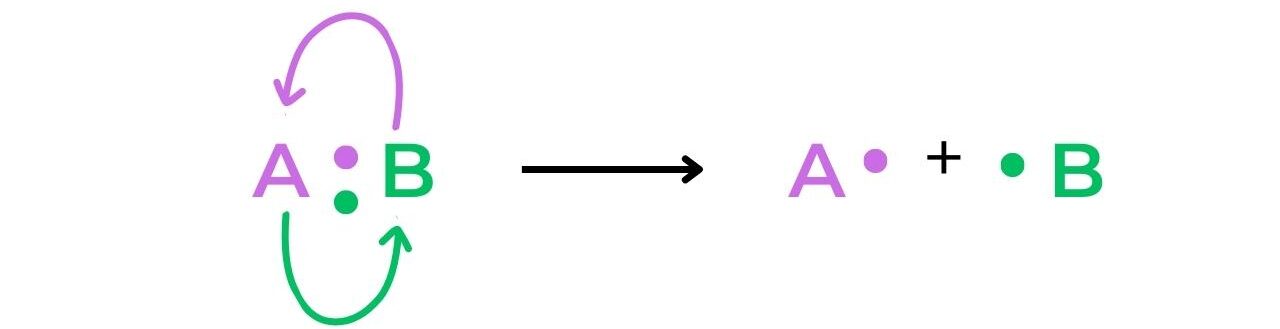
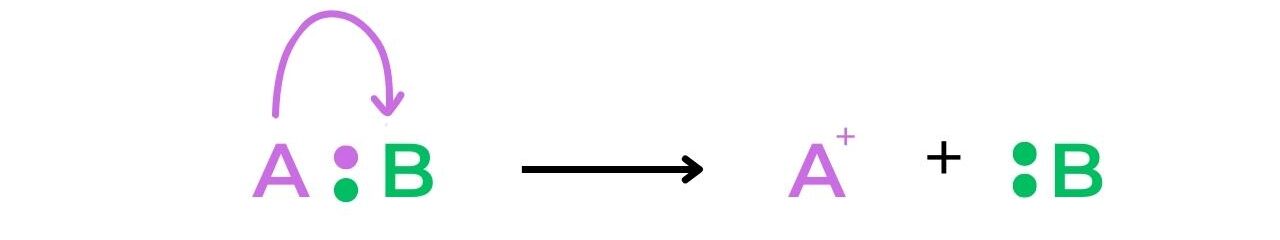
- Heterolytic fission of a bond creates a cation and an anion, as the electrons involved in the bond are unevenly split between the two atoms:
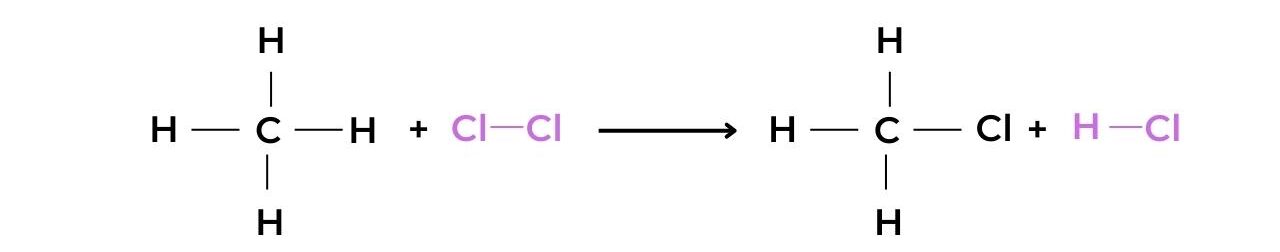
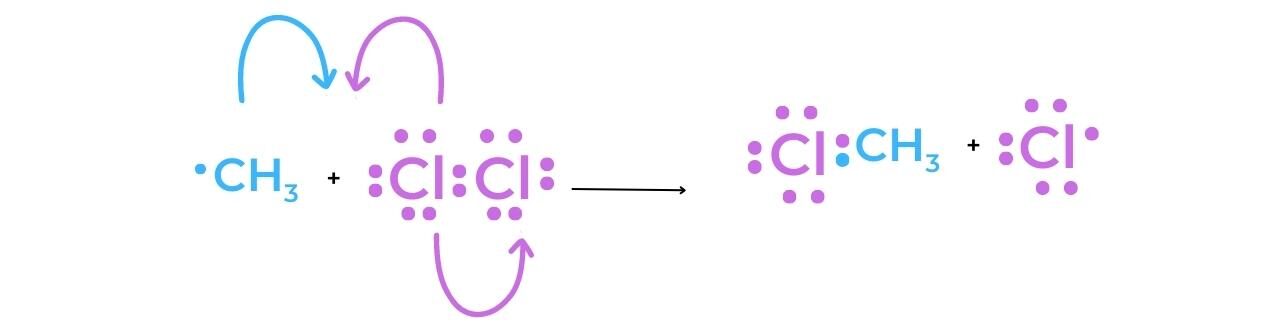
- When methane reacts with chlorine in the presence of UV light the halogenoalkane chloromethane is produced:



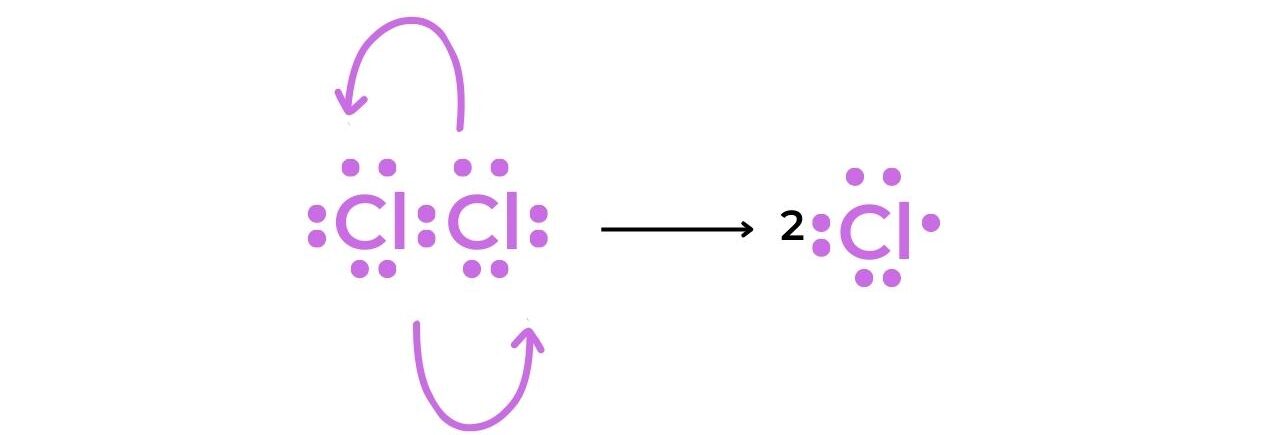
Initiation
- The homolytic fission of the chlorine molecule in the presence of UV light produces two chlorine radicals that have a short lifespan.

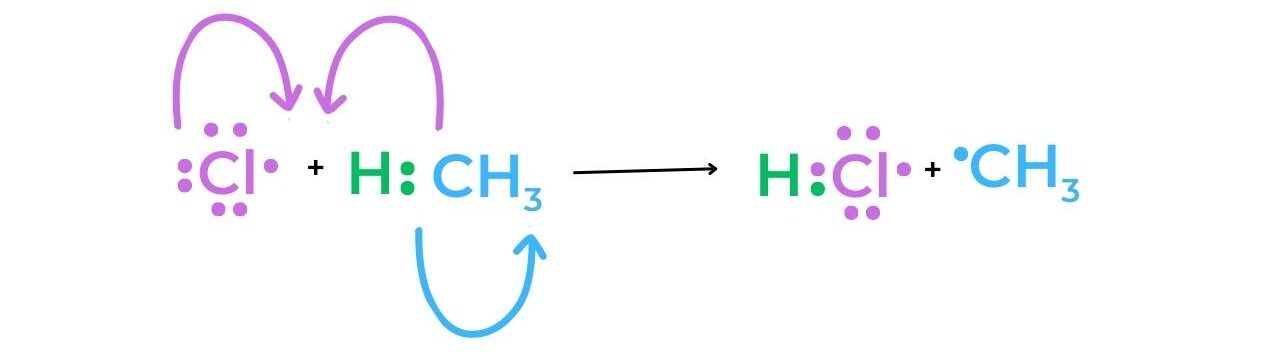
Propagation
- Methane and a chlorine free-radical undergo a reaction during the first stage of propagation.
- As a result of the methyl radical’s production, the reaction can continue as a chain reaction. The desired halogenoalkane, chloromethane, is produced when the methyl radical reacts with a chlorine molecule to produce a chlorine radical that can now participate in the first propagation reaction.


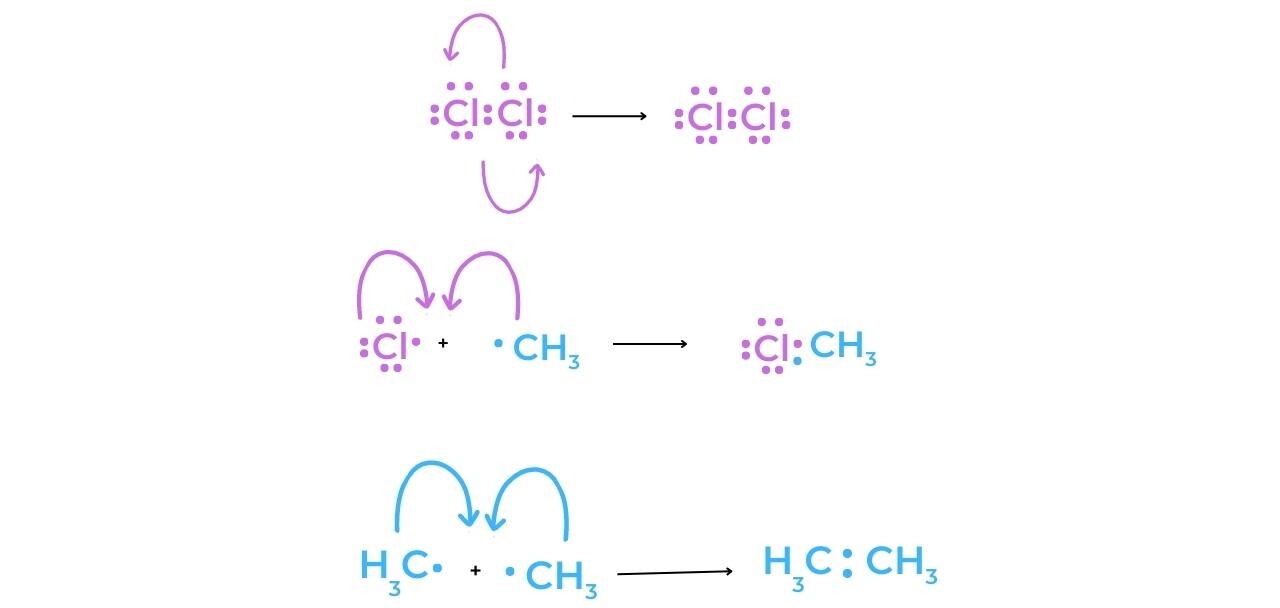
Termination
- The concentration of radicals in the reaction mixture is decreased during a termination step. As the hydrocarbon concentration begins to decrease, termination reactions become more common. They “mop up” the radicals, slowing and eventually stopping the reaction completely.

Alkenes
- Alkenes are hydrocarbons with at least one carbon–carbon double bond that are unsaturated.
- Alkenes are more reactive than their saturated counterparts due to the double bond. Alkenes undergo reactions through addition.
Test for unsaturation
- The presence of a double bond in a hydrocarbon can be demonstrated using the addition of bromine water, Br2(aq). A mixture of the alkene and bromine water will undergo a colour change from brown to colourless:
C2H4(g) + Br2(aq) → C2H4Br2(aq)
brown colourless
- The mechanism of this reaction is shown in figure 2. If there is no colour change with bromine water this is a negative result, indicating the absence of the carbon–carbon double bond.

Polymerization of alkenes
- The plastics industry is one of the largest manufacturing bodies in the world, producing a broad range of addition polymers used widely for a variety of purposes.
- Addition polymerization is the reaction of many small monomers that contain a carbon– carbon double bond, linking together to form a polymer. The ethene monomer, supplied to the plastics industry by the petrochemical industry, undergoes addition polymerization to produce the monomer polyethene:
nC2H4 → [CH2-CH2 ]n
Alcohols
- Alcohols form a diverse group of compounds that have a wide range of applications and play a significant part in synthetic reactions.
- Alcohols can undergo complete combustion, releasing carbon dioxide and water:
C2H5OH(l) + 3O2(g) → 2CO2(g) + 3H2O(g) ∆H = -1367 kJ
Primary alcohols
- The oxidation of a primary alcohol is a two-stage process that first produces an aldehyde followed by a carboxylic acid.
- Potassium dichromate(VI), K2Cr2O7 is a milder oxidizing agent than potassium manganate(VII), KMnO4. When the primary alcohol ethanol, C2H5OH is heated with acidified K2Cr2O7, the aldehyde ethanal, CH3CHO is produced. This aldehyde can be further oxidized to the carboxylic acid ethanoic acid, CH3COOH.

Secondary alcohols
- The oxidation of a secondary alcohol such as propan-2-ol results in the formation of a ketone:

- Upon formation of the ketone, no further oxidation is possible as the carbon atom of the functional group has no hydrogens attached to it.
Condensation reaction of an alcohol and a carboxylic acid
- Esters are derived from carboxylic acids and have a variety of applications ranging from flavouring agents and medications to solvents and explosives.
- Esterification is a reversible reaction that occurs when a carboxylic acid and an alcohol are heated in the presence of a catalyst, normally concentrated sulfuric acid:
- The IUPAC names of esters consist of two words. The first word is derived from the name of the alkyl chain in the alcohol (in this example, “methyl”). The second word is composed of the root name of the carboxylic acid (in this case, “prop”), followed by the suffix “anoate”.

Nucleophilic substitution reactions: An introduction
- Once an unreactive alkane has undergone a substitution reaction to form a halogenoalkane, the resulting molecule can take part in other types of reaction.
- Since halogenoalkanes contain a polar carbon–halogen bond, C-X, the electron-deficient carbon is now open to attack by electron-rich species known as nucleophiles.
- Nucleophiles contain a lone pair of electrons and sometimes carry a full negative charge. Nucleophiles act as Lewis bases.
- An example of nucleophilic substitution is the reaction of chloroethane with aqueous sodium hydroxide. This reaction produces an alcohol (ethanol) and releases a chloride ion, Cl , the leaving group
CH3CH2Cl(g) + OH (aq) → CH3CH2OH(aq) + Cl (aq)
Electrophilic substitution reactions: An introduction
- Benzene does not readily undergo addition reactions. Instead it will undergo electrophilic substitution reactions.
- An electrophile is an electron-poor species capable of accepting an electron pair. It acts as a Lewis acid.
- An example of electrophilic substitution is the reaction of benzene with elemental bromine. This reaction takes place in an anhydrous environment and requires a Lewis base (FeBr3 or AlBr3) as the catalyst.

Types of organic reactions
Nucleophilic substitution reactions
- The substitution reactions of saturated alkanes (sub-topic 10.2) involve homolytic fission, creating free-radicals that possess unpaired electrons. The formation of chloroethane from the reaction between ethane and chlorine is an example:
C2H6(g)+ Cl2(g) → C2H5Cl(g)+ HCl(g)
- The chloroalkane that is produced has very different properties from those of the alkane, and can undergo substitution reactions, producing a wide variety of compounds with different functional groups.
- The reason for this increased reactivity is the highly electronegative chlorine atom and the polar halogen–carbon bond.
- There are two types of nucleophilic substitution (SN). The mechanism of an SN reaction depends on whether the halogenoalkane is primary, secondary, or tertiary.
SN2 reactions and primary halogenoalkanes
- Nucleophilic substitution in primary halogenoalkanes proceeds in one step. The rate- determining step (slow-step) involves both the halogenoalkane and the nucleophile so the rate of reaction is dependent on the concentrations of both reactants. It is described as a second-order reaction.
rate = k[halogenoalkane][nucleophile]
- For example, the reaction between bromoethane and aqueous hydroxide ions yields ethanol and the leaving group, the bromide ion.

- In this “backside attack”, the nucleophile attacks the electrophilic centre at 180° to the position of the bromine leaving group, the large halogen atom creating steric hindrance. Such hindrance by bulky substituents prevents “frontal attack” by a nucleophile.
- As the reaction proceeds, the entering nucleophile causes an inversion of configuration, in the same way that an umbrella blown inside out in a storm. This is known as the Walden inversion. Hence, the SN2 reaction is said to be stereospecific.
- This is a reaction where starting reagents differing only in their configuration are converted into stereoisomeric products.
Drawing mechanisms for SN2 reactions
- The nucleophile’s curly arrow begins with its lone pair, or negative charge, and reaches the carbon atom.
- The curly arrow representing the bromine leaving group originates at the bond between the carbon and bromine atoms. This can be shown on bromoethane or on the transition state.
- Partial bonds, HO C Br, are represented by dotted lines.
- The transition state is enclosed in square brackets with a single negative charge.
- The formation of the product and the leaving group must be shown.
SN1 reactions and tertiary halogenoalkanes
- Tertiary halogenoalkanes undergo nucleophilic substitution reactions that involve two steps. The rate-determining step involves only the halogenoalkane: the bond to the leaving group breaks, forming a carbocation. The reaction is a first-order reaction.
rate = k[halogenoalkane]
- For example, the reaction between 2-chloro-2-methyl propane and aqueous hydroxide ion yields 2-methylpropan-2-ol and the chloride ion (figure 4). The reaction has a molecularity of one, as there is only one molecular entity involved in the elementary reaction and hence is termed unimolecular.

Drawing mechanisms for SN1 reactions
- The curly arrow representing the halogen leaving group originates at the bond between the carbon and the halogen.
- The representation of the carbocation clearly shows a positive charge centred on the carbon atom.
- The curly arrow from the nucleophile originates from its lone pair or negative charge, terminating at the carbon atom.
- The formation of the product and the leaving group must be shown.
Factors affecting the rate of nucleophilic substitution
The rate of a nucleophilic substitution reaction depends on three main factors.
1) The identity of the halogen
- The presence of a good leaving group in a reactant undergoing nucleophilic substitution increases the rate of reaction of both SN1 and SN2 mechanisms. In both cases the rate- determining step involves the heterolytic fission of the carbon–halogen bond, in which the two bonding electrons move to the more electronegative atom
- Bond strength as well as electronegativity is important in choosing a leaving group (table 2). Fluoroalkanes are virtually inert due to the short length (138 pm) and the high strength of the C F bond (492 kJ mol–1).
- As you move down the halogen group the strength of the carbon–halogen bond decreases as the size of the halogen atom increases. A larger halogen results in longer, weaker bonds.
Carbon – halogen bond | Bond enthalpy/ kJ mol-1 |
C– F | 492 |
C – Cl | 324 |
C – Br | 285 |
C– I | 228 |
Table 1: Bond dissociation energies of carbon halogen bonds
2.) The classes of halogenoalkane: Primary, secondary, or tertiary
- As we have seen, tertiary halogenoalkanes predominantly undergo nucleophilic substitution via the SN1 mechanism while primary halogenoalkanes favour the SN2 mechanism. For secondary halogenoalkanes, both mechanisms are possible (table 3).
- Because of the inductive effects of the alkyl groups that are bonded to the carbon atom, a tertiary carbocation is more stable than a primary carbocation. The more stable tertiary carbocation quickly forms following the cleavage of the carbon-halogen bond and immediately reacts with the nucleophile.
Halogenoalkane | Mechanism |
primary | SN2 |
secondary | SN2/SN1 |
tertiary | SN1 |
Table 2: Prevalence of the SN2/SN1 mechanisms in different classes of halogenoalkanes
3.) The choice of solvent
- SN2 reactions are best performed in aprotic, polar solvents while SN1 reactions are carried out in protic, polar solvents.
- Aprotic, polar solvents are suitable for SN2 reactions because they:
- possess no O-H or N-H groups so they cannot form a hydrogen bond to the nucleophile
- cannot solvate the nucleophile so leaving it “naked” and maintaining its effectiveness as a nucleophile in forming the transition state.
- Examples of aprotic solvents include ethyl ethanoate, CH3COOCH2CH3, and propanone, CH3C(O)CH3
- Protic, polar solvents are suitable for SN1 reactions because they are polar in nature due to the presence of polar bonds
- possess either an O-H or N-H group so can form hydrogen bonds with the nucleophile
- solvate the nucleophile, thus inhibiting its ability to attack electrophiles such as the δ+ carbon atom.
- Solvation is the process by which solvent molecules surround the dissolved ions. The smaller the nucleophile, the more effective the solvation. Because the nucleophile is encapsulated by the solvation shell, it is less effective as a nucleophile in forming an SN2 intermediate; therefore SN1 reactions are favoured.
What makes a good nucleophile?
- A nucleophile is an electron-rich species capable of donating a pair of electrons to an electrophile to create a covalent bond. The ease with which these electrons can be made available determines a nucleophile’s strength.In summary:
- Both nucleophiles possess at least one pair of electrons, so by definition, they can act as Lewis bases.
- The negatively charged hydroxide ion is a stronger nucleophile than the water molecule which is its conjugate acid. An electrophile is much more attracted to a negatively charged ion than to a neutral molecule.
Electrophilic addition reactions
- In contrast to a nucleophile, an electrophile is an electron-deficient species that will accept a pair of electrons, acting as a Lewis acid. Electrophiles include the nitronium ion, NO2+ and the methyl cation,CH3+
- Electrophiles have either a formal positive charge (a cation) or a partial positive charge (δ+) generated by the presence of a highly electronegative species resulting in the polarization of the bond.
- A carbon–carbon double bond contains both a sigma (σ) bond and a pi (π) bond (figure 9). A sigma bond is formed by the end-to-end overlap of atomic orbitals and electron density is centred between the nuclei of the bonding atoms, along the internuclear axis.
- A pi bond is formed by the sideways overlap of atomic orbitals and electron density found above and below the plane of the nuclei of the bonding atoms.

Markovnikov’s rule
- Markovnikov’s rule can be used to predict the main products of electrophilic addition of hydrogen halides to unsymmetrical alkenes (see below). The carbon atom of the alkene, which is already bonded to the greatest number of hydrogen substituents, will be the preferred bonding site for the hydrogen atom.
- Due to the reduction in density of the positive charge caused by the inductive effects of the three alkyl substituents, a tertiary carbocation is more stable than a primary carbocation.
Type of carbocation | 3° | 2° | 1° |
Level of stability | most stable | least stable | |
Structure |
Table 3: The relative stabilities of primary, secondary and tertiary carbocations form the basis of Markovnikov’s rule
Electrophilic addition of hydrogen halides to alkenes
- In the electrophilic addition reaction between but-1-ene and hydrogen iodide, the major product is 2-iodobutane as the 2° carbocation is formed preferentially. Hydrogen iodide is split heterolytically, creating the hydrogen cation, H+ and the iodide anion, I . The initial attack on the pi electrons of the C=C bond comes from the cation, followed by a rapid reaction between the unstable carbocation and the halogen ion (figure 10).

Drawing mechanisms for electrophilic addition reactions
- A curly arrow originates from the carbon–carbon double bond to the hydrogen atom of hydrogen iodide.
- The curly arrow representing the iodine leaving originates at the bond between the hydrogen and iodine atoms.
- A curly arrow goes from the lone pair or the negative charge on I to C+ (the carbocation).
- The structural formula of the product 2-iodobutane is shown.
Electrophilic addition of halogens to alkenes
- Often used as a test for unsaturation in organic molecules, the electrophilic addition of a halogen (specifically bromine) to an alkene follows the same mechanism as shown in figure 11.
- The halogen is a non-polar molecule with a net dipole of zero. It is polarized as it approaches the electron-rich C=C of the alkene: electrons within the halogen molecule are repelled, resulting in a temporary dipole.

Electrophilic addition of interhalogens to alkenes
- Interhalogens are molecule-to-molecule combinations of two or more halogens.Differences in electronegativity between the halogens will result in an electrophilic region of the molecule and this determines which halogen will attack the pi bond (figure 12).
- The addition reactions of halogens, interhalogens, and hydrogen halides to symmetrical alkenes all undergo the same mechanism as with unsymmetrical alkenes (figure 13). The difference is that Markovnikov’s rule does not apply.
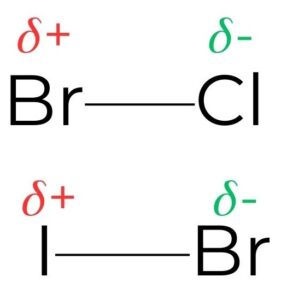

Electrophilic substitution reactions
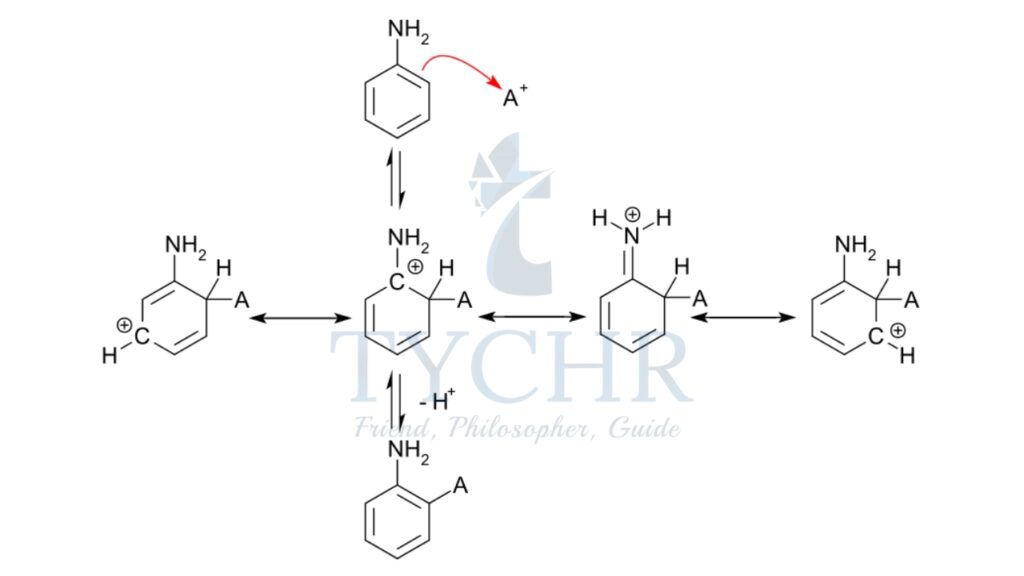
- The electrophilic substitution mechanism of these reactions can be illustrated by the nitration of benzene.
- The electrophile nitronium ion NO2+ must be produced for the first step. A nitrating mixture of sulfuric acid and nitric acid at 50 °C produces a higher concentration of nitronium ions, allowing the reaction to proceed at an acceptable rate. Pure nitric acid only contains a small amount of this electrophile. Nitric acid is protonated by sulfuric acid, which then lets go of a water molecule to produce the electrophile.:
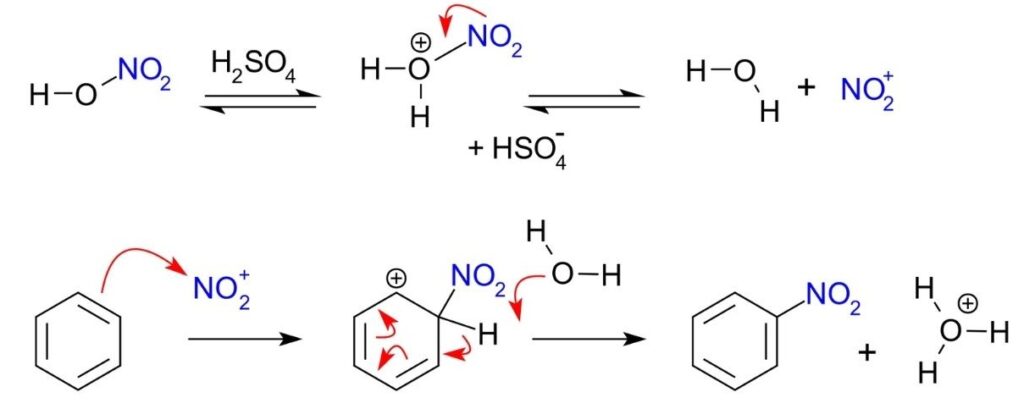
- The nitronium ion is a strong electrophile. As the electrophile approaches the delocalized pi electrons of the benzene ring, the nitronium ion is attracted to the ring. Two electrons from the ring are donated and form a new C N bond. Additionally, a pi electron from the N=O bond of the nitronium ion moves onto the oxygen atom:
- This is the rate-determining step of the mechanism. The addition of the nitronium ion to the C=C bond eliminates the aromaticity of the arene.
- Water then acts as a base, deprotonating the carbocation intermediate and restoring the aromaticity of the system.
- The product nitrobenzene is a yellow oil that can be isolated from the reaction mixture.


Drawing mechanisms for electrophilic substitution reactions
- A curly arrow originates from delocalized electrons in benzene and terminates at the +NO2 electrophile.
- Structural representation of the carbocation shows a partial circle and a positive charge on the ring.
- A curly arrow representing the hydrogen ion leaving originates at the bond between the carbon and hydrogen atoms and terminates at the benzene ring cation.
- The structural formula of the organic product nitrobenzene is shown along with the released hydrogen ion, H+.
Reduction of carboxylic acids
- Carboxylic acids are reduced to aldehydes and eventually to primary alcohols while ketones are reduced to secondary alcohols, in reactions that are the reverse of the oxidation of alcohols.
- The reduction equation is often represented in a simplified manner using the symbol [H] to represent the reducing agent. The reduction of an aldehyde to a primary alcohol can be represented by the general equation:
R-CHO + 2[H] → R-CH2OH
- and the reduction of ketones to secondary alcohols by this general equation:
R-CO-R′ + 2[H] → R-CH(OH)-R′
Synthetic routes
Background to designing a synthetic route
- Organic synthesis takes a starting material and converts it via a series of reactions into the desired product.
- Each step produces an intermediate in quantities less than the theoretical yield, so an efficient synthetic pathway will involve the smallest number of steps.
- Reactions that convert one functional group to another, such as the oxidation of a primary alcohol to a carboxylic acid or the nucleophilic substitution of a halogenoalkane, do not change the length of the carbon chain.
- Synthetic tools include controlled chain-lengthening and chain-shortening reactions while polymerization involves the formation of long molecules made up of repeating monomer units.

Retro-synthesis
- Knowledge of the types of reactions undergone by functional groups and their mechanisms allows chemists to determine possible steps in a synthetic pathway, in both the forward and the reverse directions.
- Step 1: Draw the structural formulae of both the starting compound and the desired product(s). Identify the functional group(s) present in the product.
- Step 2: List possible reactions that would produce the desired functional group(s).
- Step 3: Identify the functional group(s) present in the starting material and identify any relationship between the starting reagent and any intermediate compounds you have listed in step 2.
- Step 4: Design a reaction pathway that has the minimum number of steps. Include all the reaction conditions and reagents required.
Stereoisomerism
Types of isomerism
- Stereoisomers have an identical molecular formula and bond multiplicity but show different spatial arrangements of the atoms. Stereoisomers can be subdivided into two major classes, conformational isomers and configurational isomers.
- Conformational isomers can be interconverted by rotation about the σ bond, without breaking any bonds. Configurational isomers can only be interconverted by the breaking of the σ or π bond or through rearrangement of the stereocentres.
Flowchart 1: Different types of isomers.
Conformational isomers
- Substituents and functional groups joined together by single σ-bonds can rotate freely, changing the three-dimensional arrangement of the atoms relative to one another. In contrast, a carbon–carbon double bond is composed of both a σ and a π bond and the arrangement of electron density above and below the internuclear axis means that no rotation is possible without breaking the π bond.
- As a result, conformational isomers differ from one another in how their atoms are arranged around a single bond. The rapid interconversion between conformers makes it nearly impossible to separate the individual isomers..
Configurational isomers
- Configurational isomers can be interconverted only by the breaking of bonds or through the rearrangement of the stereocenters. Configurational isomers are subdivided into cis–trans and E/Z isomers on the one hand and optical isomers on the other.
- As mentioned above, configurational isomers exist due to the lack of rotation around the carbon–carbon double bond such as that present in aliphatic alkenes.
- Cis–trans isomers are determined by the positions of substituents relative to a reference plane. For alkenes this reference plane is the carbon–carbon double bond. Cis-isomers have substituents on the same side of the reference plane while in trans-isomers the substituents are on opposite sides (figure 8).

Optical isomerism
- Optical isomerism is a type of configurational isomerism determined by the presence of chiral carbon atoms.
- Also known as a stereocentre or asymmetric centre, a chiral carbon is bonded to four different atoms or groups of atoms.
- Optical isomers have the ability to rotate plane-polarized light and exist in pairs that are called enantiomers or diastereomers.



- Enantiomers are non-superimposable mirror images of each other. They have no plane of symmetry and their optical activity is most readily assigned when the molecules are represented as three-dimensional images (figure 13).

Optical isomers and plane-polarized light
- Under the same conditions, two optical isomers with the same general formula rotate the plane of polarized light by the same angle but in opposite directions (figure).
- One enantiomer rotates the plane of polarization in a clockwise direction; this is designated the (+) enantiomer.
- The other enantiomer rotates the plane of polarization in an anticlockwise direction and is designated the (-) enantiomer.
- A 50:50 mixture of the two enantiomers is called a racemic mixture (or racemate) and does not rotate plane-polarized light.
- The optical purity of synthetic reaction products can be measured with a polarimeter.
- This technique is commonplace in industry producing optically active products, examples of which can be found throughout the pharmaceutical, fragrance, food, and chemical industries.
- The product’s effect on plane-polarized light can be compared with literature values to determine the purity of the desired enantiomer.

Diastereomers
- Diastereomers are distinct from optical isomer enantiomers. They are not superimposable, like enantiomers, but they do not form mirror images.
- They differ in the arrangement of at least one center and have two or more stereocenters. Diastereomers with the same general formula have distinct physical and chemical properties, in contrast to enantiomers.
- In option B.10 D-xylose and L-ribose are identified as diastereomers due to the configuration at the C-2 and C-4 positions (figure 19).

are non-superimposable but they are not mirror image
